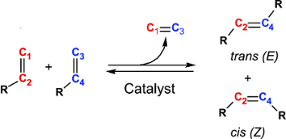
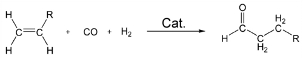
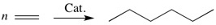ResearchResearch interestsThe main goal of most of our ongoing research projects is to develop new and improved transition-metal-based catalysts. Molecular-level computational chemistry is an important tool in order to reach this goal. The initial role of theory in these projects usually is to provide mechanistic information, meaning that the most efficient catalyst development should be based on a clear understanding of the underlying factors governing the activity or selectivity. However, theory can provide more than just the mechanism: Due to decades of development in both computational methodology and hardware, it has become cost-efficient to move large portions of the catalyst screening-work from the lab to the computer, and integration of computational chemistry into catalyst development is an important part of our work. The predicted new catalysts are subsequently synthesized and tested, either in our own lab or in the labs of collaborating groups. Depending on the chemical problem in question, the methods used in the computational parts of these projects range from molecular mechanics through semi-empiry and density functional theory to high level, wave function-correlated ab initio methods. The use of more approximate methods is routinely preceded by validation against experimental data or results from higher level calculations. Main current research activitiesIndustrial catalysisMost of our activity is within the downstream application of natural gas, with a particular focus on the use of olefins as the main substrate in a range of refinement processes:
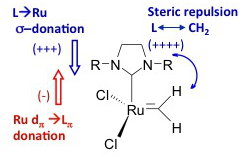
Method developmentWe develop methods and approaches for design and development of catalysts and other functional transition-metal compounds. We have, for example, worked on incorporating chemically intuitive and meaningful molecular descriptors, such as measures of ligand-to-metal donation, metal-to-ligand back-donation, and steric repulsion, into quantitative structure-activity relationship (QSAR) models. The result has been powerful and very predictive models from which it is also straightforward to extract chemical information and understanding. The limitation of this approach is that it is manual in the sense that a human being still has to suggest, build and subject the candidate catalyst structure to calculations to obtain a computational measure of its suitability as a catalyst. We are thus, together with the group of Prof. Bjørn K. Alsberg, NTNU-Trondheim, working to automate the procedure of designing new catalysts and other functional transition-metal compounds. We develop a de novo in silico design method based on an evolutionary algorithm in which the fitness (or cost function) is obtained from quantum chemical or other molecular-level calculations. Overview of the development of the approaches and methods towards design:
|